The Role of the N terminal region of the Flagellar Switch protein FliG
A thesis presented to the Department of Molecular Biophysics and Biochemistry, Yale University, in Candidacy for the Degrees of B.S./M.S. May 1998
By Stacey Denenberg
Research Supervisor: Dr. Robert Mcnab.
Abstract
Salmonella typhimurium
is a motile bacterium propelled by a rotating "tail," termed flagellum The flagellar motor, which creates the rotation, is composed of many proteins, each with distinct tasks within the mot or. FIiG, FILM, and FIiN form what is called the "switch complex," which is responsible for the alternation between clockwise and counterclockwise motion. Of the three proteins, FliG, which is located between the MS and the C ring of the flagellum is most involved with the actual rotation of the flagella. To study the roles of different regions in FliG, a series of FIiG mutant proteins, each with a 10 amino acid deletion, were created by PCR mutagenesis. These deletions began with amino acid 7 and continu ed through the N terminal portion of the protein After confirming that the resultant mutant proteins were stable, plasmids containing the mutant genes were transformed into a FliG null strain. The resultant transformants were studied for flagellati on and motility in both liquid medium and on soft agar motility plates. The results revealed that the first three deleted regions completely destroyed flagellation and motility while the next four deleted regions had varying effects on flagellation and mo tility. The deletion mutants were also transformed into a wild-type strain to study dominance effects of the mutant FIiG protein on the wild-type motor. Subsequently, I cloned the deletion mutant genes into a vector that allowed overexpression of the FIiG mutant proteins. I then purified FliF protein, the main component of the MS ring in the flagellar motor. After confirming previous results suggesting that FliF binds to a FIiG fragment containing only the first 108 amine acids, I performed affinity blots on the FliG deletion mutants. The first six deletion mutants did not interact with FliF while the next four deletions interacted with FliF as well as the wild-type FliG, suggesting that the amino acids essential for FliG’s interaction with FliF fell with in the first 66 amino acids of FliG’s N terminus.Introduction
Both E. coli and Salmonella typhimurium are motile bacteria propelled by a rotating "tail," termed flagella. It is composed of a long, external helical filament, a hook, and a series of rings mounted on a central rod that anchors the flagellum in the cell envelope (DePamphilis and Adler, 1971). The flagellar motor creates the torque needed to rotate the flagellum through a transmembrane proton motive force, and is comprised of many proteins, each with distinct tasks within the motor.
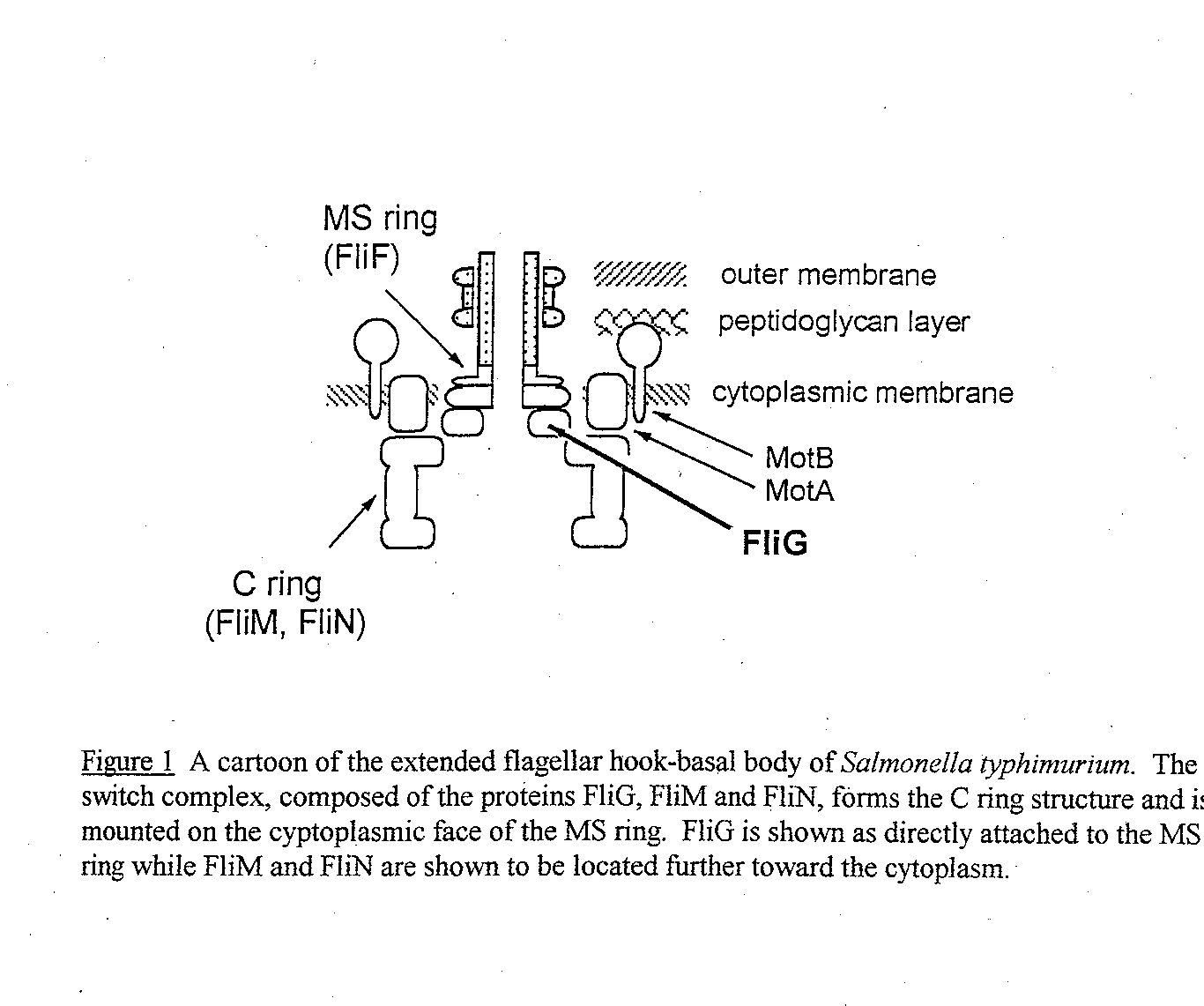
FliG, a 36,800 Dalton protein, is located on the cytoplasmic face of the cell membrane between the MS and the C ring of the flagellar motor (see Figure 1) (.Francis et al, 1992). Along with the proteins FliM and FIiN, it forms the "switch comple x," which is responsible for the alternation between clockwise (CW) and counterclockwise (CCW) motion. During CCW rotation, the filaments bundle together and propel the cell forward in what is called "smooth swimming." During CW rotation the filaments com e apart, causing the cell to "tumble," randomly reorienting the cell (Macnab, 1995).
In recent times, many studies have attempted to distinguish separate functions for the three proteins, FIiG, FliM and FliN. Spontaneous mutagenesis studies supported the hypothesis that FIiG was primarily involved with the rotation of the flagella (Iri kura et al, 1993; Sockett et al, 1992). That is, many spontaneous mutations created paralyzed and null phenotypes instead of "switch biased" phenotypes. Based on these observations, it seemed logical to create sets of deletions in the FIiG p rotein and study the resulting phenotypes. A ten amino acid deletion proved large enough in a previous study of FIiM to ensure that any remaining function could be attributed to a different region of the protein (Toker et al, 1996). Therefore, in C hapter I, I created 7 successive ten amino acid deletions using a PCR mutagenesis strategy. The deletions began with amino acid 7 and covered the N terminal portion of FliG has previously been shown to interact with FliF, another protein in the flagellar motor (Francis et al, 1992). FliF is a 62,000 Dalton protein that is the major component of the MS ring. It was shown that a spontaneous mutation causing an in-frame FIiF-FliG fusion protein was motile and underwent switching, indicating that FliG and FliF must be stationary in relation to each other(Francis et al, 1992). Francis et al also found another spontaneous FliF-FliG fusion protein in which the C terminal 56 amino acids of FIiF and the N terminal 94 amino acids of FIiG were d eleted. This deletion fusion mutant was partially motile, indicating that FliG’s N terminal region interacts with FIiF. Also, according to electron micrographs of hook basal bodies (HBBs) that had been decorated with gold-protein A, FliG is associated wit h the HBB even when the C ring is not present (Francis et al, 1994). In Chapter II, I attempted to better define the region of FliG that interacts with FliF. To obtain more definitive results, I cloned the deletion mutants into a pET vector, which allowed for protein overexpression and further probing using affinity blotting. I confirmed the results obtained by Brian Lane in 1995 by performing an affinity blot using purified FliF protein and two FliG fragments. The fragment containing the N termina l 108 amino acids interacted with FIiF while the fragment containing the rest of FliG did not interact with FliF, giving further evidence that FliG’s N terminal region interacts with FIiF. I then proceeded to narrow down the region of interaction by perfo rming affinity blots with the FliG deletion mutant proteins.
Chapter I - FIiG Deletion Mutant Creation and Characterization
Materials and Methods
Bacterial Strains and Plasmids
E. coli strain DFB225 was a gift from David Blair (University of Utah) and has been characterized as an in-frame null deletion of FUG (Lloyd et at, 1996). RP437 is the parent strain of DFB225 and is wild-type for motility and chemotaxis (Parkinson and Houts, 1982). XL1-Blue cells (Stratagene; La Jolla, CA) were used for cloning experiments and Western blotting growths. pUC18 (IBI; New Haven, CT), which confers ampicillin resistance to its host cells, was used as a cloning vector. pUC19G, containing the wild-type FliG gene of S. typhimurium, cloned into the BamHI and HindlII sites, was used as the positive control (Anne Toker, personal communication). Plasmid pAMH3 (Kihara et al., 1989) contains flagellar regio n IIIb of S. typhimurium and was used as a template for PCR mutagenesis. Mutants were named pax, where x is a number 1 through 7. The first deletion starts at amino acid 7 at the N terminus and the other deletions follow consecutively.
Media
Luria broth and plates were prepared according to Miller et al., 1972. Soft tryptone motility agar plates (1% Tryptone, 0.7% NaC1, 0.35% Bacto agar) were prepared according to Armstrong and Adler, 1967.
Construction of FliG Deletion Mutants
The following procedure to create 10 amino acid deletions was formulated by Anne Toker (Toker et al, 1996). All primers were synthesized using a Model 393 DNA/RNA Synthesizer (Applied Biosystems, Foster City, CA). The mutagenic PCR proce dure is shown in Figure 2. Primer SFliF-XbaA was complementary to a region in thefts gene upstream of FliG, and created an XbaI site at the 5’ end of the PCR fragment. Primer SFliH-HindB was complementary to a region in FliH downstrea m of FliG and created a HindllI site at the 3’ end of the fragment. A series of mutagenic primers (denoted mutA and mutB in Figure 2) were used to delete 30 base pair regions of the FliG gene, thereby deleting 10 amino acids in the FI iG protein.
All mutA primers were exactly complementary to the sequence immediately downstream of the deleted region. Each mutB primer’s 3’ region was complementary to the sequence immediately upstream of the deleted region while the first 12 base pairs were compl ementary to the 5’ end of the corresponding mutA primer. First, PCR reactions using pAMH3 as a template and performed in a MJ MiniCycler (MJ Research, Watertown, MA) created two fragments: a fragment upstream of the deletion created with primers SFIiF-Xba A and mutB, and a fragment downstream of the deletion created with SFliH-HindB and mutA. The fragments were visualized on a 0.7% TAE agarose gel, purified using the QIAquick Gel Extraction Kit (Qiagen Inc., Chatsworth, CA), and mixed together for a second round of PCR. This PCR; using the two outside primers SFliF-XbaA and SFliH-HindB, created the final fragment containing the 10 amino acid deletion.
Cloning and Purification of Deletion Mutant Plasmids
The PCR fragments (purified as above) containing the FliG deletions were cloned by digesting each fragment with XbaI and HindlII and ligating them into pUC t 8 digested with the same two restriction enzymes. The ligated pla smids (e.g. pal, pA2, etc.) were then transformed into XL1-Blue competent cells and plated on Luria plates containing ampicillin, isopropylq3-D- thiogalactopyranoside (1PTG), and X-galactose (X-Gal). White colonies were selected, grown overnight in 1.5 ml Luria broth with ampicillin at 37°C, and the plasmids were harvested using the QIAprep Spin Plasmid Miniprep Kit (Qiagen). The correct plasmids were identified by two methods. First, the plasmids were digested with XbaI and HindllI and run on a 0. 7% TBE agarose gel to ensure that the insert was present and was the correct size. Second, all clones were sequenced following the protocol of the Sequenase Kit (US Biochemicals; Cleveland, OH). Each deletion mutant clone was sequenced in the area immedia tely surrounding the deletion to ensure that no mutations had occurred in the region around the primers. At least three independent clones for each deletion were used in the following experiment to rule out anomalous results.
Swarm Plate Assays
Mutant and control plasmids transformed into either DFB225 or RP437 cells were spotted onto soft tryptone agar motility plates containing 50 gg/ml ampicillin. After incubating at 30°C for 22 hours, the swarms were compared to cells transformed with wild type FliG because the size of the swarm is usually proportional to the motility of the cells. Motile cells will swim out radially, creating a "swarm" following a chemotactic gradient of nutrients (see Figure 3), while nonmotile cells will remain at the point of the original innoculum. Colonies that appeared null, or to have no swarm, were studied under the dissecting microscope to look for the presence of small trails or other changes not visible to the naked eye.
Motility in Liquid Medium
Mutant and control plasmids transformed into DFB225 or RP437 cells were grown in Luria broth containing 50 µg/ml ampicillin at 30°C for approximately 7 hours to achieve optimal motility (checked microscopically). The cells were then diluted 1:1 0 into motility medium (10 mM PO4, 0.1 mM EDTA, pH 7) and observed under the high intensity dark-field microscope (Macnab, 1976) to determine both level of flagellation and motility relative to wild-type RP437 cells. The motility of each mutant was estima ted as a percentage relative to wild-type motility.
Western Blotting
Western blots were performed to determine the level of the mutant protein in XL 1-Blue cells. Samples were grown at 30°C with vigorous shaking until the cells reached optimal flagellation and motility. Optical density measurements (at) = 600 r an) were used to standardize the amount of protein in each sample. The cells were centrifuged, washed once, and resuspended in 100 µl of protein sample buffer. The samples were boiled for 5 minutes before loading a 12.5% SDS-PAGE mini-gel (Hoefer Mighty S mall; San Francisco, CA) and transferred to a nitrocellulose membrane for 2 hours at 70 volts using a Hoefer Transblot. After gentle shaking in 5% milk in Tris buffered saline plus 0.1% Triton X-100 (TBS-T) for 2 hours, the membranes were washed with TBS- T and incubated with a 1:10,000 dilution of polyclonal anti- FIiG (a generous gift of K. Oosawa and S.-I. Aizawa, Teikyo University, Japan). After several washes, the membrane was incubated with a 1:10,000 dilution of goat anti-rabbit horseradish peroxida se (HRP) (Bio-Rad; Hercules, CA). The antibodies were detected by enhanced chemiluminescence using the ECL detection protocol (Amersham International, UK). Coomassie gels were run in parallel to ensure that the amount of protein loaded in each lane were u niform.
Results
Mutant Phenotypes
Using PCR mutagenesis, I created seven successive 10 amino acid deletions in the N terminal region of FliG. Deletion mutant A1 deleted amino acids 7 - 16, allowing FliS’s start codon and next five amino acids to remain intact for all of the mutants. The other six mutants, named 3‘2 through A7, created successive 10 am/no acid deletions following A1. To test the complementation ability of each deletion mutant, the mutant plasmids were transformed into DFB225, a FliG null strain. A positive control, containing wild-type FliG, and a negative control ofpUC18, were also transformed into DFB225. The motility of the cells was characterized in two ways. First, swarm tests were performed. Colonies of each mutant as well as both controls, w ere spotted onto soft agar motility plates containing ampicillin and incubated at 30°C for 22 hours. The results are shown in Figure 3. Mutants A1, A2, and A3 appeared completely null, while mutants A4 through A7 increased in motility stepwise with 3,7 ap proaching wild-type swarming. Mutants 3,1, 3,2, and A3 were also studied under the dissecting microscope to check for small trails indicating the presence of a few motile cells, but none were apparent. These results indicate that deleting the first 30 ami no acids destroys motor function, while deleting the next 40 amino acids only impairs motor function. This implies that the first three deleted regions are essential either for the proper folding of FliG or for its interaction with the other motor protein s. Because the next four deletion mutants are motile, they must sustain the correct interactions, although with varying efficiency compared to wild-type FliG. Second, the deletion mutants were studied in liquid medium. After transforming each mutant plasm id plus the positive and negative controls into DFB225, individual colonies were grown in Luria broth for 7 hours at 30°C for optimal motility and flagellation. Cell phenotypes were then studied using both low-intensity microscopy and high-intensity dark field microscopy. The results are shown in Table 1. Using low-intensity microscopy, rankings were given for cell motility, with + denoting poor motility to +++ denoting wild-type motility. These results are in approximate agreement with the swarm tests. A ll motile cells exhibited both tumbling and smooth swimming, indicating that the switch between CW and CCW rotation was not biased in one direction. High-intensity dark field microscopy, which permits the visualization of individual flagellar filaments, w as used to study the level of flagellation of each mutant. The nonmotile mutants were not flagellated, or a fla’ phenotype, indicating that the deletion affected the assembly of the flagellar filaments. Deletion mutants 3,4 through 3,7 were flagellated, i ndicating that those deletions did not block flagellar formation. But, these last four deletions did affect motor finaction to varying degrees, as only 3,7 was close to wild-type motility.
Table 1 - Phenotypes under Low and High-Intensity Microscopy
|
Mutant |
Flagellated |
# Flagella |
Motility |
Tumbling |
Smooth Swimming |
|
pa1 |
No |
0 |
None |
-- |
-- |
|
pA2 |
No |
0 |
None |
-- |
-- |
|
pA3 |
No |
0 |
None |
-- |
-- |
|
pA4 |
Yes |
4 |
+ |
Yes |
Yes |
|
pA5 |
Yes |
4-6 |
++ |
Yes |
Yes |
|
PA6 |
Yes |
4-6 |
++ |
Yes |
Yes |
|
PA7 |
yse |
6 |
+++ |
Yes |
Yes |
Table 1 Mutants were grown from a single colony in Luria Broth plus ampicillin for approximately 7 hours for optimal motility. The information in the first two columns was determined by high-intensity dark field microscopy, while the information in the final three columns was determined by low-intensity microscopy. For the motility column, cells were ranked by none, indicating that only Brownian motion was present, +, indicating Poor motility, ++, indicating moderate motility, and +++, indicatin g close to wild-type motility. The number of flagella for each mutant is the average of many different cells studied.
Test for Dominance Effects
Dominance tests were performed to study the effect of the deletion mutant proteins in a wild-type background. The deletion mutant plasmids were transformed into RP437, and tested for both swarming ability on soft tryptone motility plates and for motili ty in liquid medium. Although swarming ability and motility in liquid culture was affected to varying degrees for the different mutants, no dominance effect was observed at the level of flagellation. That is, all transformants had flagellation levels simi lar to those of the wild-type RP437 cell. The swarming ability of each mutant, as shown in Figure 4, is ranked 0 through 10, with 0 indicating a null (non-motile) phenotype and 10 denoting wild-type swarming. RP437 transformed with pUC 19G swarmed as well as cells transformed with just pUC 18, indicating that excess wild4ype FIiG does not affect cellular motility. None of the deletion mutant swarms were as large as either control, indicating that all of the mutant proteins affected motility at least sligh tly. The swarms get smaller from A1 to A7, indicating that deletions further into FIiG are interfering more with the wild-type strain’s swarming ability. Dominance effects were also apparent in tests of the deletion mutants in liquid medium. Motility in l iquid medium was also ranked on a scale of 0 to 10 as before (Figure 4). Wild-type cells transformed with pUC 19G were just as motile as cells transformed with pUC 18, confirming the previous observation that excess levels of FliG do not affect motility. Also, none of the deletion mutants allowed the cells to swim as well as wild-type cells. There seemed to be no apparent correlation, however, on the position of the deletion, and the dominance effect in liquid medium. Comparing the swarm tests to the liqu id motility tests shows some discrepancies in dominance effects. A smaller dominance effect during swarm tests could be explained by the existence of selective pressure for motile cells on soft tryptone plates that does not exist in liquid media. Mutants A5, A6 and A7, however show a much stronger dominance effect on swarm tests, because a high proportion of cells were motile in liquid medium but the cells swarmed very poorly on motility plates. A possible explanation could be that the interaction between wild-type and mutant FliG reduced the efficiency of the flagellar motor. Therefore, a high portion of the cells would be flagellated and look motile in liquid medium, but the cells would be sluggish and therefore swarm more slowly than expected.
Western Blots
Western blots were performed to check the cellular levels of the mutant FliG protein. These results, shown in Figure 5, were very specific, with almost no background bands. All seven deletion mutants had bands as intense as the positive control , with mutants A2, A3 and A4 actually having slightly more intense bands. Coomassie gels run in parallel confirmed that the amount of protein loaded in each lane was the same (data not shown). This experiment indicates that all seven of the deletion mutan ts synthesize at least as much stable protein as wild4ype FIiG from a pUC vector, and suggests that the mutant phenotypes were not caused by a lack of stable FIiG protein in the cells. Western blots comparing the chromosomal expression level of FliG in wi ld4ype RP437 to the pUC19G expression level revealed that there is a much higher level of FliG protein produced by the plasmid than produced by the chromosomal copy of the gene (data not shown).
RP437 cells did not produce enough FIiG to create an intense band, while the positive control and the deletion mutants caused intense bands, indicating that the level of plasmid protein production is much higher even without induction by IPTG. This is not surprising, because pUC is a high copy plasmid. The increased level of FliG protein does not seem to affect the cells, as evidenced by the previous dominance test results (see Figure 4).
Discussion
This study can be broken into three parts. First, I created seven deletion mutants spanning the N terminus of FliG using PCR mutagenesis and confirmed the stability of the mutant proteins through immunoblotting. Second, I characterized the mutant pheno types in a null background using both swarm tests and liquid motility tests. Finally, I characterized the mutant phenotypes in a wild-type background, looking for dominance effects. This study builds on previous ones, such as Francis et al, 1992 an d Lane, 1995. The results suggest a lot about the role of FliG’s N-terminus in the flagellar motor, but careful assumptions must be made in order to correctly interpret the data collected
The first part of this study involved creating the deletion mutants. The act of creating the deletion mutants itself provided little information, but the final constructs were useful in further experiments. The complementation tests provided evidence f or the role of FliG’s N terminus, when taken in the correct context. A ten amino acid deletion could cause severe damage to the protein, so I assumed that any function surviving the deletion is probably mediated by another region of FliG. For instance, no ne of the deletion mutants created a switch-biased phenotype, indicating that the region of FliG involved with motor switching functions was not affected. A non-flagellate phenotype, however, would provide less clear-cut information. That is, I was not ab le to simply assume that the deletion in a nonflagellate sample was important solely for flagellar assembly.
Plasmids containing wild-type FliG were able to restore mobility to a ris null strain. This implies that deletion mutants which failed to restore function could have had a number of problems, such as a significantly disrupted structure or a fail ure to make important interactions with other proteins, but the possibility that a copy of FliG transcribed from a plasmid was not able to complement a chromosomal deficiency can be ruled out.
It is possible that some of the deletions disrupted the three dimensional struc����������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������nt forms of FIiG. There are approximately 26 subunits of FliG per basal body (Irikura et al, 1993), and it would be reasonable to assume that they interact with each other within the flagellar motor. Therefore, FliG must interact with itself efficiently, or a strong dominance effect would be seen.
While the first three deletion mutants may be essential to either protein folding or flagellation (or possibly both), the last four deletion mutants provide a bigger clue as to the role of the N terminal region of FIiG. Because there is no switch bias, other regions of FliG probably control the switch. Instead, deletion mutants A4 to A6 cause the flagellar motor to rotate less efficiently. This could just mean that the proteins are misfolded, or it could mean that the deleted regions are important (but not essential) for interactions with other proteins involved in flagellar rotation. It has long been known that FliG interacts strongly with the cytoplasmic face of the MS ring (Francis et al, 1992), which is constructed with subunits of FliF prot ein (see Figure 1). FIiF and the MS ring is essential for flagellar assembly, and previous studies have shown that the N terminal region of FliG interacts with FliF (Francis et al, 1992; Lane, 1995). The previous studies narrowed FliG’s region of i nteraction with FUF to a region of about 100 amino acids, but based on the complementation results, I hypothesize that the region of interaction may be smaller. That is, deletion mutant 7 seems to restore full motility to a mdl strain, so it does not see m essential for any interactions. In Part II of this study I use the deletion mutants to examine the interaction between FliF and FliG by performing affinity blots.
Chapter H - FIiG Interactions with FIiF
Materials and Methods
Bacterial Strains and Plasmids
XL1 Blue cells (Stratagene) were used for all cloning experiments. E. coli BL21 (DE3) cells were used for the overexpression of genes cloned into pET vectors through the T7 overexpression system (Studlet et al., 1990). The FIiG deleti on mutants created in Chapter I were supplemented with other deletion mutants created by the same protocol. These deletion mutants were called pA8 through pA32 and contained 10 amino acid deletions immediately following pA7 (May Macnab, personal communica tion). Deletion mutants pal through pal0 were digested out of pUC18 using XbaI and HindIII restriction enzymes and then ligated into the vector pET22b (Novagen; Madison, WI) which had been digested with the same two enzymes. The FIiG deletion mutan ts are now called pETAx, where x is a number 1 through 10. Other FIiG fragments were cloned into pET 11 a (Novagen; Madison, WI) and are as follows: pBL2000, wild- type FliG, pBL2100, containing amino acids 1 to 108 of FliG, and pBL2200, containing amino acids 109 to 331 of FliG (Lane, 1995). The plasmid pFFF1300, a pET His-Flag-FliF clone, was used for FliF overexpression (Fan Fan, personal communication).
Overexpression.
The FIiG deletion mutants in pET1 la or pET22b were transformed into BL21 (DE3) and grown in Luria broth plus ampicillin (50 ulcerating/ml) at 37°C to an O1)6oo = 0.60. 1 mM IPTG was added and the cells were shaken at 37°C for an additional 2.5 hou rs.. 0.50D of cells were pelleted by centrifugation and resuspended in 100 I, µl of protein sample buffer. 10 [µl aliquots were boiled for 5 minutes and mn** on a 12.5% SDS-PAGE gel.
FliF Purification.
The following protocol is an adaptation of a protocol originally created by Fan Fan (personal communication). The plasmid pFFF1300 was transformed into BL2I (DE3) and grown in 1 L Luria broth plus ampicillin at 37°C to an OD600 = 0.5. The cells wer e induced with 1 mM IPTG, grown for an additional 3 hours, spun down in 250 ml aliquots and frozen at -20°C for later use. One aliquot of cells was resuspended in 10 ml binding buffer (20 mM Tris-HC1, pH 7.9, 0.5 M NaCI, 5 mM imidazole), incubated for 0.5 hours with 25 mg lysozyme at 4°C, and sonicated. After addition of Triton X-100 to 1%, the cells were centrifuged. All of the following steps were performed at 4°C. The supernatant was loaded onto a 3 ml Ni-NTA column (Qiagen) and rotated for 2 hours. Th e column was washed successively with binding buffer plus 15 mM imidazole at 1% Triton X-100 followed by 0.2% Triton X-100. FIiF was eluted using strip buffer (100 mM EDTA, 0.1% Triton X-100) and collected in 1.5 ml fractions. The fractions were visualize d using Coomassie staining on a 12.5% SDS-PAGE gel, and the fractions containing pure FliF were Pooled and dialyzed overnight against 20 mM Tris plus 0.1% Triton X-100. The concentration of FliF was determined by the Bradford assay protocol (Bio- Rad; Her cules, CA), using bovine serum albumin to create the standard curve. FliF was stored in 4 mg aliquots at -80°C.
To identify the most specific way of probing for FliF, western blots were performed on 10 µl aliquots of the purified FliF mixed with 2 µl of protein sample buffer (see below for the full western blot protocol). Both anti-FLAG (Hopp et al, 1988) (IBI; New Haven, CT) and monoclonal anti-FliF (Homma et al, 1987)) were tested as the primary antibody. As shown in Figure 6, monoclonal anti-FliF provides a much more specific interaction and was thus used for all affinity blotting experiments. < /P>
Western Blotting and Affinity Blotting.
Western blotting was performed to confirm the expression of the FliG deletion mutants in the T7 overexpression system. The FliG deletion mutants in pET22b and FliG variants in pET1 la were transferred from 12.5% SDS PAGE gel to nitrocellulose membr ane by electrocution as in Chapter I and probed with anti-FliG.. As can be seen in Figure 7, the expression of the deletion mutants in the pET system was comparable to the expression when the deletion mutants were in pUC18. Also shown in Figure 7 is a wes tern blot of the FIiG variants in pET1 la. The fact that the protein expressed from pBL2100 is faint or broad can be explained by its extremely low molecular weight of approximately 6,000 Daltons. That is, the band did not resolve as well as the other, hi gher molecular weight, deletion mutants. Affinity blotting was performed as follows. After the FliG variants were transferred to nitrocellulose membrane by electroelution, the membrane was blocked with 5% milk in TBS-T for 1.5 hours and then incubated ove rnight with 0.4 gg/ml of purified FliF at room temperature with shaking. The membrane was washed and then incubated with a 1:10,000 dilution of monoelonal FliF antibody, incubated with a 1:3,000 dilution of goat anti-mouse HRP (Bio-Rad), and developed fol lowing the protocol of the ECL Western Blotting detection kit.
Results
Preliminary Affinity Blot
To study the interaction between FliG and FIiF, I first confirmed previous results indicating that FliF would bind to a fragment containing only the first 108 amino acids of FliG (Lane, 1995). After performing a Western blot of the FliG fragmen ts to confirm a detectable level of protein (see Figure 6), I performed an affinity blot using 0.4 gg/ml purified FliF protein. As expected, the fragment containing the first 108 amino acids of FliG interacted with FIiF while the fragment containing amino acids 109 to 331 did not interact with FliF (Figure 8). This experiment served two purposes. First, it confirmed the previous results. Second, it served as a positive control for a slightly modified affinity blot protocol.
Plasmid Construction
Preliminary experiments with affinity blots used FIiG in a pUC vector, because the deletion mutants created in Chapter I were in pUC18. Wild-type FliG expressed from a pUC vector did not produce an interaction with FliF in an affinity blot (dat a not shown). I hypothesized that FliG would need to be overexpressed in order for this experiment to work. Therefore, the FIiG deletion mutant genes (A1 through A10) were recloned into pET22b. pET vectors can be induced using IPTG in a T7 expression syst em, creating a much higher expression level of the desired protein. Deletion mutants A8 through A10 were included because the previous result suggested that the essential amino acids fell within the first 108 amino acids, and I wanted to test that entire region.
Affinity Blots
After confirming that protein expressed from the pET vectors was stable (see Figure 7), I performed affinity blots using the deletion mutant proteins. As shown in Figure 9, deletion mutants pETAl through pETA6 did not interact with FliF while delet ion mutants pETA7 through pETAl0 interacted with FliF. This suggests that the amino acids deleted in pETA7 through pETAl0 are not essential for the interaction between FliG and FliF. Further, it is likely that all of the amino acids essential for the inte raction between FliG and FliF fall within the first 66 amino acids of FliG’s N terminus (remembering that the first six amino acids were never deleted).
Discussion
To interpret the information provided by the affinity blots, I first had to make a few assumptions. Western blots showed detectable amounts of each deletion mutant, so I felt safe in assuming that the deletion mutants were stable in the cell. It is not necessarily true, however, that the deletion mutants are folded into FliG’s native structure. A principal limitation of any Western or affinity blot is that the protein is denatured in SDS during electrophoresis before being transferred to the nitroc ellulose membrane. One hopes that the protein refolds into its native form during the washes, but it is difficult to confirm that the protein has in fact refolded correctly.
Another problem inherent in the experiment is that deleting ten amino acids may strongly affect the folding of the protein. The protein may fold well enough to prevent degradation in the cell, but the altered conformation may block the deletion mutant’ s interaction with FIiF. Therefore, the absence of an interaction with a specific deletion mutant may be caused by a change in the protein folding instead of an actual deletion of amino acids essential for the interaction with FliF.
It should also be noted that Figures 8 and 9 contain background bands. This does not, however, detract from the validity of the experiment. FliF is known to interact with other proteins in the flagellar motor, so it is not surprising that there are oth er bands present in the blot. The background bands are fainter than the potential deletion mutant band because the other proteins are present in much lower (chromosomal level) concentrations. These background bands stay constant throughout the positive an d negative controls, so I feel that the background bands can be discounted. Also, Figure 6 shows that monoclonal anti-FliF is very
References
Armstrong, J.B. and Adler, J. (1967) Chemotaxis in Bacteria. Annu. Rev. Biochem. 44, 341- 356.
DePamphilis, M.L and Adler, J. (1971) Fine Structure and Isolation of the Hook-Basal Body Complex of Flagella from Escherichia coil and Bacillus subtilits. d.. Bacteriol. 105, 384-395.
Francis, Noreen R., Irikum, Vera, Yamaguchi, Shigem, DeRosier, David J., and Macnab, Robert M. (1992) Localization of the Salmonella typhimurium flagellar switch protein FliG to the cytoplasmic M-ring face of the basal body. Proc. Natl. Acad. Sci. USA 89, 6304-6308.
Francis, Noreen R., Sosinsky, Gina E., Thomas, Dennis, and DeRosier, David J. (1994) Isolation, Characterization, and Structure of Bacterial Flagellar Motors Containing the Switch Complex. J. Mol. Biol. 235, 1261-1270.
Homma, M., Aizawa, S.I., Dean, G.E., and Macnab, R.M. (1987) Identification of the M-ring Protein of the Flagellar Motor of Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 84, 7483-7487.
Hopp, T.P., Pricket, K.S., Price, V.L., Libby, R.T., March, C.J., Cerretti, D.P., Urdal, D.L., and Conlon, P.J. (1988) A short polypeptide marker sequence useful for recombinant protein identification and purification. Bio/Technology. 6, 1204-12 10.
Irikura, V.M., Kihara, May, Yamaguchi, S., Sockett, H, and Macnab, R.M. (1993) Salmonella typhimurium FIiG and FliN Mutations Causing Defects in Assembly, Rotation, and Switching of the Flagellar Motor. J. Bacteriology. 175, 802-810.
Kihara, May, Homma, M., Kutsukake, K., and Macnab, R.M. (1989) Flagellar Switch of Salmonella typhimurium: Gene Sequences and Deduced Protein Sequences. J. Bacteriol. 171,
3247-3257.
Lane, Brian. (1995) Deletion Analysis of the Flagellar Switch Protein FliG and Investigation of the Interaction Between FliG and the MS Ring Protein FIiF by Affinity Blotting.
Lloyd, Scott A., Tang, Hua, Wang, Xun, Billings, Stephanie, and Blair, David F.(1996) Torque Generation in the Flagellar Motor of Escherichia coB: Evidence of a Direct Role for FliG but Not for HiM or FliN. J. of Bacteriology. 178, 223-23 1.
Macnab, Robert M.(1976) Examination of Bacterial Flagellation by Dark-Field Microscopy. J. Clin. MicroBiol. 4, 258-265.
Macnab, Robert M. (1995) "Flagellar Switch." In: Two-Component Signal Transduction. J.A. Hoch and T.J. Silhavy, eds. Pp181-199. American Society for Microbiology. Washington, DC.
Miller, J. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 1972.
Parkinson, John S. and Houts, Susan E.(1982) Isolation and Behavior of Escherichia colt Deletion Mutants lacking Cherootaxis Functions. J. of Bacteriology. 151, 106-113.
Sockett, H., Yamaguchi, S., Kihara, May, trikura, V.M., and Macnab, R.M.(1992) Molecular Analysis of the Flagellar Switch Protein FliM of Salmonella typhimurium. J. Bacteriol. 174, 793-806.
Studier, F.W., Rosenberg, A.H., Dunn, J.J., and Dubendorff, J.W.(1990) Use ofT7 RNA Polymerase to Direct Expression of Cloned Genes. Methods Enzymol. 185, 60-89.
Toker, Anne S., Kihara, May, and Macnab, Robert M.(1996) Deletion Analysis of the FIiM Flagellar Switch Protein of Salmonella typhimurium. J. of Bacteriology. 178, 7069-7079.
Tables and Figures
Figure 1 Cartoon of the Extended Hook Basal Body
Figure 2 Schematic of PCR Mutagenesis Protocol
Figure 3 Swarm Tests of Deletion Mutants in a FliG Null Strain
Table 1 Summary of Liquid Motility Tests in a FliG Null Strain
Figure 4 Dominance Effects on Swarm Plates and in Liquid Media
Figure 5 Western Blots of Deletion Mutants
Figure 6 Western Blots of Purified FliF
Figure 7 Western Blots of Deletion Mutants Expressed from pET22b
Figure 8 Affinity Blot of FliG Fragments
Figure 9 Affinity Blot of Deletion Mutants Expressed from pET22b

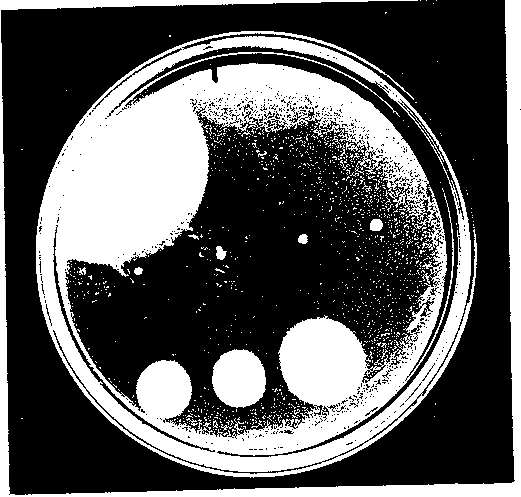
Figure 3 A representative swarm plate of the deletion mutants in DFB225. The plates were incubated at 300C for 22 hours. The top row (L to R), pUC19G (positive control) and pUC18 (negative control). The middle row (L to R), deletion mutants A1, A2, A3 and A4. The bottom row (L to R), deletion mutants A5, A6 and A7.
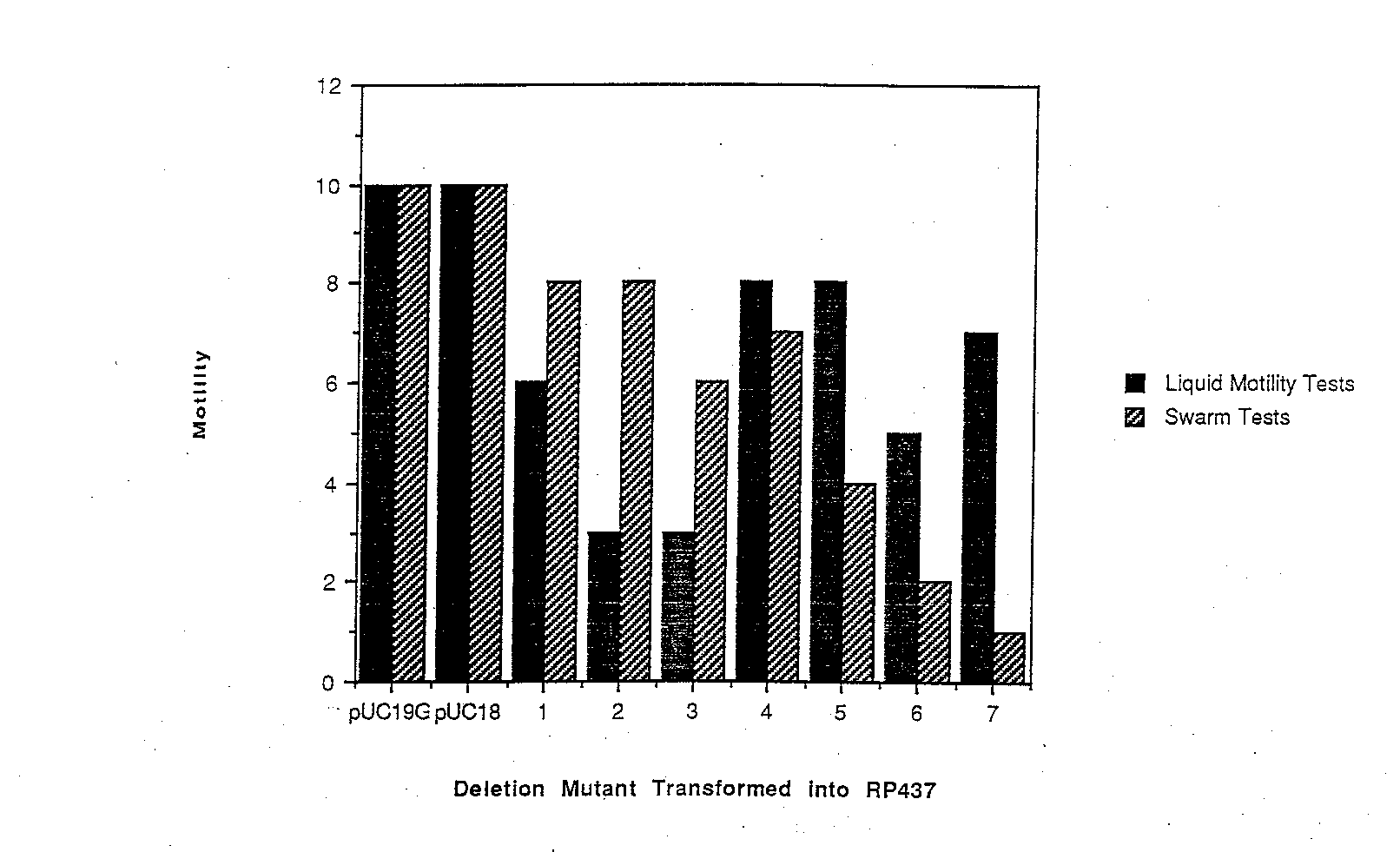
Figure 4 A summary of dominance effects measured by swarming ability and motility in liquid medium. Each deletion mutant plasmid, as well as pUC19G and pUC18, were tested in RP437, a wild-type strain. Cell motility was ranked on a scale of 0 to 10, with 0 indicating non-motile cells and I0 indicating samples with motility similar to wild-type ceils. While excess wild-type FliG did not cause any dominance effect, the deletion mutants had varying effects, and the effects differed between swarm tes ts and liquid motility tests.
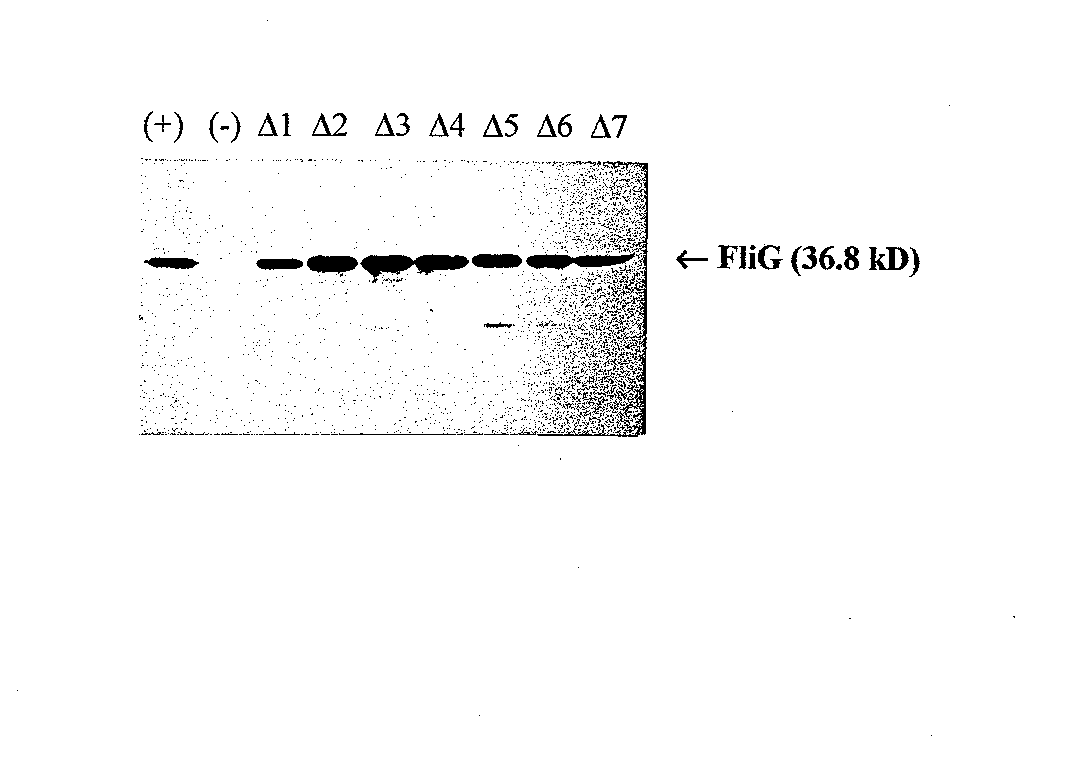
Figure 5 Western blot using anti-FliG. Lane 1,wild-type FliG; Lane 2, pUC18 (negative control); Lanes 3-9, mutants A1 through A7, respectively.
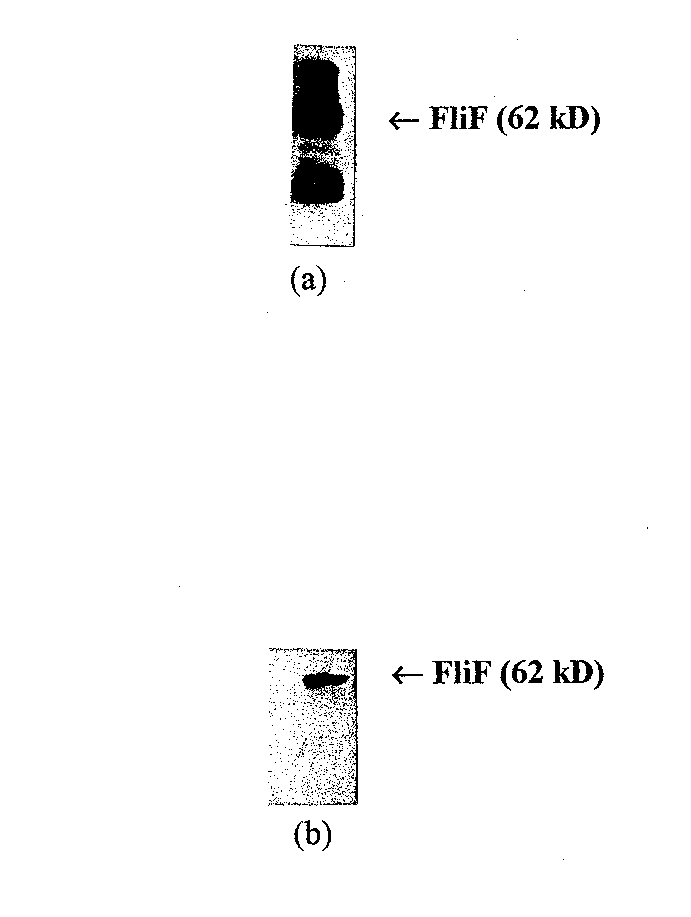
Figure 6 Western blots of purified FliF with different primary antibodies. (a) western blot with anti-FLAG directed against the FLAG epitope added to the N terminus of the FIiF protein. This blot has many nonspecific ba nds. (b) Western blot using monoclonal anti-FliF. Anti-FliF gives a much more specific result, and was chosen for affinity blotting experiments.
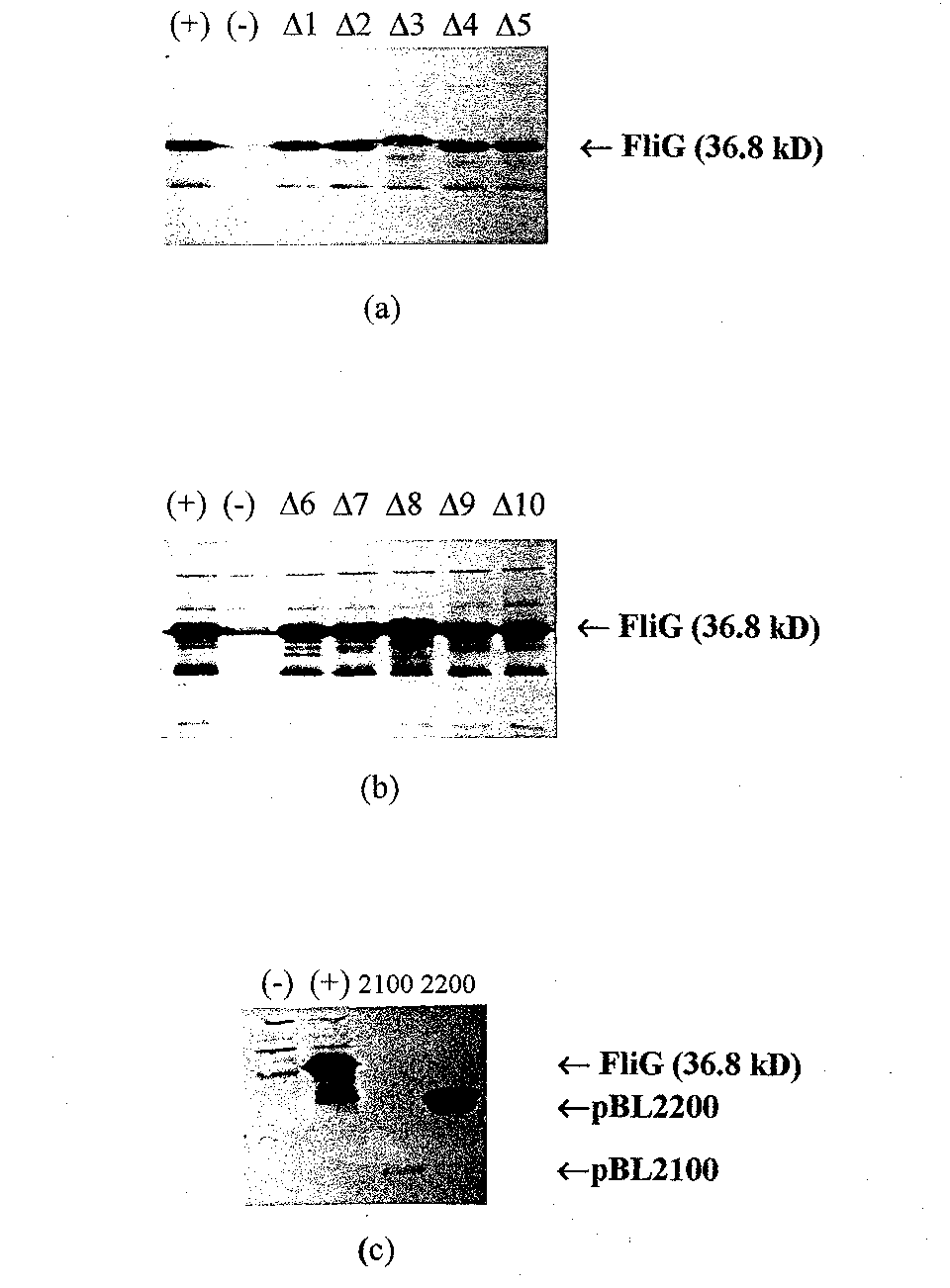
Figure 7 Western blots of the different FliG deletion mutants in pET vectors. (a) Lane 1, wild- type FliG,; Lane 2; pET22b; and Lanes 3 through 7, deletion mutants pETAl through pETA5, respectively. In (b) the first two lanes are the same as in (a), an d Lanes 3 through 7, pETA6 through pETAl0, respectively. Expression of the deletion routants seems comparable to expression ofw/ld-type FliG from pET22b in both (a) and (b). (c) western blot of the FliG fragments in pET1 la. Lane 1, pET1 la; Lane 2 pBL200 0; Lane 3, pBL2100; and Lane 4, pBL2200.
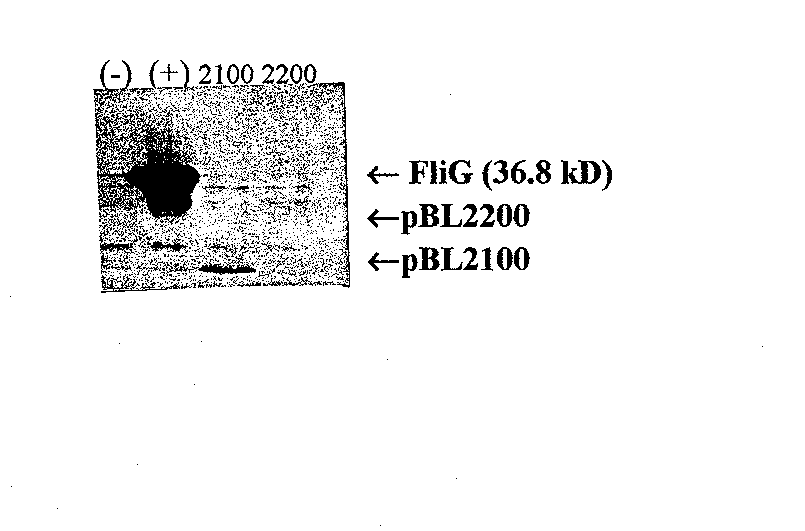
Figure 8 Affinity blot of FliG fragments probed with purified FliF. Monoclonal anti-FliF was the primary antibody. Lane l, pET1 la as a negative control; Lane 2, pBL2000 (wild-type FliG); Lane 3, pBL2100 (amino acids 1 to 108); and Lane 4, pBL2200 (amino acids 109 to 331). pBL2100 produces a positive result while pBL2200 does not, indicating that the am/no acids essential for FliG's interaction with FliF fall within the first 108 amino acids.